SQLITE NOT INSTALLED
О работе с любовью
В обычной частной клинике у метро врач просто тонет в рутине. Он может миллион лет совершать одни и те же ошибки, и никто, включая него, об этом не будет знать: консилиумов-то, собраний, каких-то клинических разборов обычно в таких клиниках нет. При этом, несмотря на лечение, дети будут выздоравливать, они вообще очень устойчивые и организмы у них крепкие, сами справляются с большинством болезней. Если с ними ничего не делать, почти всегда болезнь пройдет. Может, она будет проходить чуть дольше, может, чуть тяжелее. Но врач, который перестал учиться, никогда не узнает, прошла болезнь сама или помогло лечение. А чтобы не задумываться об этом совсем, можно сразу назначить лечение помощнее. Например, антибиотики при вирусной инфекции — на всякий случай.
Я бы не смог продержаться в такой клинике, где просто зарабатывают деньги. Я бы нигде не смог продержаться на рутинном приеме долгое время. К счастью, мне повезло, и у нас в GMS руководство много предпринимает для повышения нашей квалификации. Мы собираемся, разрабатываем внутренние протоколы, читаем друг другу лекции. И у нас есть рутина с соплями и поносами, но есть еще и Центр врожденной патологии, который привносит в работу особый смысл.
Люди выгорают, становятся грубыми, если занимаются только рутиной. Все эти врачи в поликлиниках хамят не потому, что они злые, а потому, что они ужасно устали. Родители часто ведут себя неадекватно, это нормально, когда твой ребенок болеет. И если у врача нет таких шикарных условий, как у меня здесь, если на прием отводится не час, а 10–15 минут, это приводит к злобе и агрессии: вникать нет времени, проще создать атмосферу, когда сами родители от тебя ничего хотеть не будут. Мне сложно давать рекомендации молодым врачам: как работать с любовью. Мне просто повезло.
О плюсах частной медицины
Мой приход в больницу совпал с периодом серьезных изменений, которые почти полностью отразились на клинике негативно. Незадолго до того, как мое родное грудное отделение закрыли совсем, друг позвал меня работать в Европейский медицинский центр. И я попал в совершенно новый медицинский мир. Я, конечно, знал о доказательной медицине и пытался самообразовываться в ее духе, но на это не хватало времени и ресурсов. В частной клинике впервые у меня появился платный доступ к зарубежным сайтам и, главное, время, чтобы их изучать. Этот опыт сильно изменил мое медицинское мировоззрение.
Кроме того, частная медицина открывает новые возможности для оказания более качественной помощи: нет зависимости от набора лекарств, закупаемых больницей, от тех специалистов и методов лечения, которые есть в лечебном учреждении. В платную клинику на консультацию ты можешь пригласить самого хорошего специалиста, а не того, который случайно оказался с тобой в одном штате. Здесь больше возможностей использовать препараты офф-лейбл (для лечения болезней, которые не указаны в показаниях в инструкции — прим. ред.), идти новыми путями в лечении. В государственной медицине до последнего будут использовать однажды установленные стандарты.
О миссии
В Европейском медицинском центре я познакомился с совершенно удивительным человеком — генетиком-эндокринологом, профессором Натальей Александровной Беловой. Она крупнейший специалист по врожденным нарушениям скелета и, в частности, несовершенному остеогенезу (наследственной болезни, характеризующейся ломкостью костей — прим. ред.). У Натальи Александровны таких детей наблюдалось, возможно, больше, чем у кого бы то ни было в мире. Я тоже стал заниматься несовершенным остеогенезом, и у моей работы появился абсолютно новый смысл.
Мы создали Центр врожденной патологии, специализирующийся на помощи детям с тяжелыми хроническими инвалидизирующими заболеваниями, в первую очередь с костной патологией и задержкой развития разной природы. Стратегия развития ЕМЦ не подразумевала работы с такими тяжелыми детьми, но зато мы нашли понимание и желание развивать такую сложную область в частной клинике GMS, куда центр и переехал в полном составе.
Обычно к нам приходят после путешествия по самым разным клиникам и врачам, с множеством диагнозов, часто не имеющих отношения к действительности. Чаще всего дети недообследованы, но иногда диагноз стоит верный, и вся наша помощь заключается в подборе правильной (и отмены неправильной) терапии и реабилитации. Для решения этих вопросов мы собираем консилиумы из замечательных специалистов, которых мы долго по разным местам отбирали и продолжаем отбирать. Детей с хрупкими костями мы берем на регулярное лечение в свой стационар, детей с задержками развития госпитализируем на 1–3 дня для полного обследования. Если нужно, мы можем в итоге пристроить таких детей в Центр лечебной педагогики, хорошие отделения государственных больниц, отправить за границу, а также к нашим партнерам, которые помогут в юридических и финансовых делах. То есть мы подходим к этому комплексно и стараемся своих пациентов после консультации не бросать. Часто лечение оплачивают благотворительные фонды. Когда родители к нам обращаются, мы им объясняем, как и куда для этого подавать документы.
В Центре врожденной патологии я занимаюсь организацией, участвую в консилиумах и являюсь лечащим врачом всех детей с несовершенным остеогенезом. Хрупкие кости — первая специализация нашего центра и на данный момент самая отлаженная. На консилиумах я отвечаю за педиатрические аспекты: от запоров до вакцинации. У нас в стране бытует мнение, что дети-инвалиды должны отводиться от прививок, хотя чаще наоборот, они должны прививаться активнее: для них опаснее болеть.
Диагностика
У новорождённых хромосомный анализ может быть проведён для определения точной причины врождённых дефектов.
Возможны пренатальные исследования для трисомии 18:
- Измерение уровня альфафетопротеина. Тест проводится между 15 и 17 неделями беременности. Положительный результат исследования не означает, что у ребёнка будет трисомия 18 или какая-либо хромосомная аномалия. Фактически, только около у 11% тех женщин с положительным результатом на трисомию 18 в этом тесте, действительно будет пострадавший плод.
- Ультразвук — ещё один широко используемый скрининг-тест. Как и выше описанное исследование, простое УЗИ не может быть использовано для установления диагноза трисомии 18. Более подробное исследование при помощи ультразвуковых волн может быть выполнено для поиска характерных признаков аномалии, но этот метод не может подтвердить наличие синдрома.
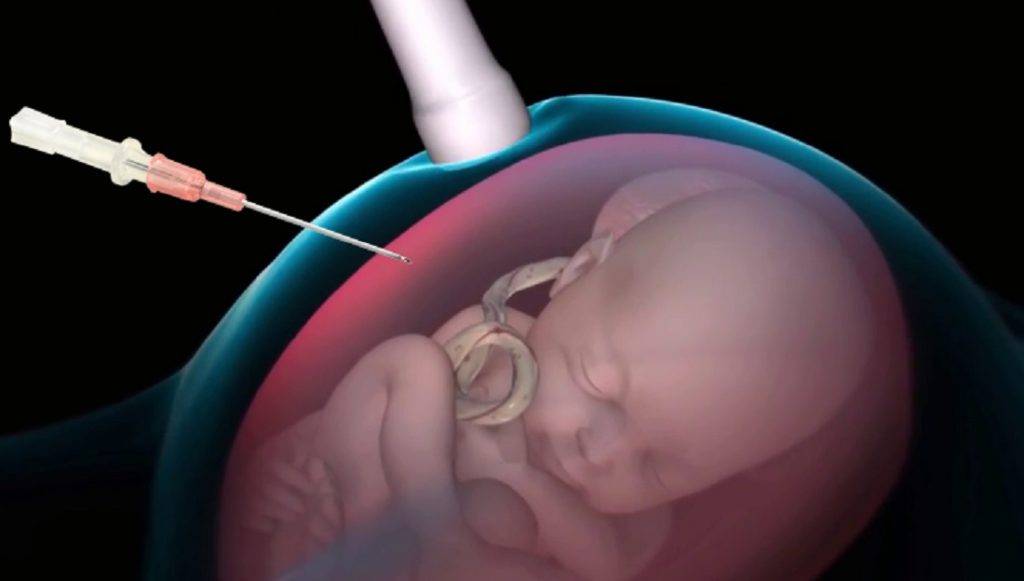
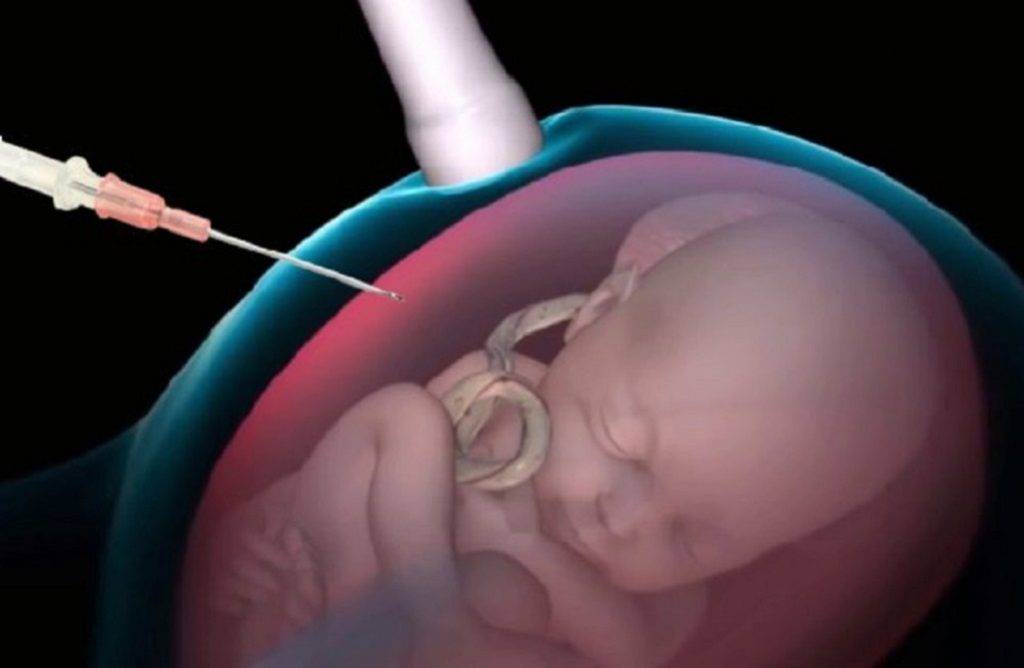
- Анализ эмбрионального хромосомного материала, полученного при амниоцентезе или биопсии ворсин хорион, необходим, чтобы доказать, что имеется дополнительная копия хромосомы 18. Амниоцентез обычно проводится на 15 — 18 неделе беременности и является наиболее часто используемым тестом для пренатальной диагностики трисомии 18. Во время этой процедуры через брюшную стенку вводят тонкую иглу и берут небольшой образец амниотической жидкости. Биопсия ворсин хориона является ещё одним типом исследования, который позволяет учить генетический материал плода. Тест выполняется через 10 — 12 недель после последнего менструального цикла и, следовательно, имеет преимущество, позволяющее установить более ранний диагноз. Эта процедура включает в себя сбор образца ворсин хориона из плаценты с помощью прокола брюшной стенки, либо с использованием катетера через влагалище.
Как проявляется синдром Патау?
Поскольку дополнительная хромосома присутствует во всем теле, трисомия 13 может вызвать проблемы во многих системах организма.
Некоторые симптомы синдрома можно лечить медикаментозно или с помощью хирургического вмешательства, но другие не поддаются лечению. Признаки включают:
- Преждевременные роды: многие беременности, при которых у плода имеется трисомия 13, заканчиваются выкидышем или мертворождением. Младенцы, которые рождаются живыми, обычно появляются на свет рано, со средним сроком гестации 29 недель. Этим детям приходится бороться со многими осложнениями и симптомами трисомии 13.
- Нарушения на лице: многие дети с трисомией 13 рождаются с расщелиной губы и/или неба. Глаза бывают расположены друг к другу вплотную и могут соединяться, образуя один глаз. Уши могут находиться низко, распространена аплазия кожи на голове (отсутствие кожного покрова).
- Нарушения сердца. Могут быть обнаружены отверстия между сердечными камерами (дефект межжелудочковой и межпредсердной перегородки) и открытый артериальный проток.
- Проблемы в мозге: у некоторых детей с трисомией 13 передняя часть не разделена должным образом. Это обуславливает многие проблемы лица, связанные с расстройством. Дети с синдромом Патау имеют серьёзные умственные недостатки, у них возможны приступы.
- Желудочно-кишечные проблемы у детей с трисомией 13 включают в себя пупочные и паховые грыжи.
- Проблемы со скелетом: у детей с синдромом могут быть дополнительные пальцы на конечностях, сжатые руки или деформированные ноги.
- Проблемы с дыханием: возможно его затруднение. Также бывают периоды, когда дети прекращают дышать (апноэ).

Причины и факторы риска
Конечно, невозможно чётко установить истинную причину, потому что у каждого ребёнка есть индивидуальные черты.
Часто заболевание происходит под влиянием нескольких факторов:
- простуда различной этиологии (атипичная пневмония, грипп, аденовирус, корь);
- реакция на вдыхаемые элементы. Это аллергические компоненты обстановки (новая мебель, все лаки, краска, новые приборы из некачественного пластика), перхоть животных, пыль. В практике большинство педиатров встречаются с семейным ларингитом, когда семья переехала в квартиру, где был произведён ремонт или замена мебели;
- анатомическая и физиологическая структура дыхательной системы (узкое горло и носоглотки). Всякое воспаление респираторной системы вызывает набухание (отёк) тканей, и гортань значительно сужается у ребёнка, что осложняет проходимость воздуха;
- сниженное качество воздуха, где находится ребёнок (горячий сухой воздух, пыль, выхлопные газы, прокуренные комнаты);
- механические факторы — повреждения гортани, перенапряжение голоса при пении, крике, вследствие громкого долгого разговора;
- гастроэзофагеальная рефлюксная болезнь и аспирация инородного тела может вызвать ларингит.
Патологии плода, вызванные алкогольной зависимостью беременной
Самая распространённая патология, вызванная употреблением алкоголя беременной женщиной, — синдром Миллера-Дикера. Мутация происходит в гене 17-й хромосомы. Главная причина аномалии — интоксикация плода альдегидами, передающимися через материнскую кровь.
На УЗИ синдром Миллера-Дикера выражается в многоводии, отставании внутриутробного развития плода и снижении его двигательной активности. На более поздних сроках можно увидеть утолщение коры головного мозга при разглаживании мозговых извилин.
Голова у больных детей меньше положенного размера, лоб выпуклый, плоский затылок, челюсть недоразвита (алкогольная дизморфия). Ушные раковины расположены ниже положенного уровня, пальцы неправильной формы, тазобедренные суставы находятся на зачаточном уровне, стопы укорочены, задний проход сросшийся.
Такие дети обычно умирают в возрасте в 2 лет из-за аспирационной пневмонии. У них глубокая умственная отсталость и отсутствуют даже обычные рефлексы (глотание, моргание).
Проявления заболевания
Младенец с синдромом Патау. Аринэнцефалия с циклопией.
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
При синдроме Патау наблюдаются тяжёлые врождённые пороки. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У них выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. У 80 % новорождённых встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезёнки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.
В связи с тяжёлыми врождёнными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).
Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных синдромом Патау до 5 лет (около 15 % детей) и даже до 10 лет (2 — 3 % детей).
Оставшиеся в живых страдают глубокой идиотией.
Другие синдромы врождённых пороков развития (синдромы Меккеля и Мора, тригоноцефалия Опитца) по отдельным признакам совпадают с синдромом Патау. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Признаки Синдрома Патау
- низкая масса тела при доношенной беременности (менее 2500 грамм);
- неправильное строение черепа (небольшие размеры, сужение лба, расширение затылочной области);Выраженные отклонения физического и умственного развития;
- пороки развития структур головного мозга;
- пороки развития глаз (отсутствие глазных яблок, маленькие глаза, катаракта, отслойка сетчатки и др.);
- заячья губа;
- волчья пасть;
- деформированная форма ушей;
- пороки развития кисти (лишние пальцы, неправильное формирование большого пальца);
- локальное отсутствие кожи, волос;
- деформированные стопы, лишние пальцы на ногах;
- множественные пороки мочевыводящей, сердечно-сосудистой, пищеварительной и половой систем.
Диагностика
Пренатальный скрининг на синдром Патау
В первом триместре врач будет проводить обычные скрининговые тесты, такие, как ультразвуковое исследование и анализ крови матери. Результаты этих тестов предоставят специалисту информацию о том, как растёт и развивается ребёнок. Например, во время процедуры УЗИ врач может заметить аномалии в физическом облике ребёнка, которые могут указывать на трисомию 13.
Если специалист полагает, что у плода возможно хромосомное расстройство, он может предложить матери провести неинвазивный пренатальный генетический скрининговый тест. Это исследование предоставит врачу информацию о риске наличия у плода трисомии 13 или иной хромосомной аномалии. Если результаты этого скринингового теста показывают высокий риск, беременной женщине будет предложено провести диагностическое тестирование.
Пренатальное диагностическое тестирование синдрома Патау
Пренатальные диагностические тесты предоставят специалисту окончательный ответ относительно того, имеет ли ребёнок определённую хромосомную аномалию. Пренатальные диагностические варианты тестирования для синдрома Патау включают процедуры:
- Обследование хорионических ворсинок. Исследование обычно проводится между 10 — 13 неделями гестации. Эта процедура подразумевает взятие образца плацентарных хорионических ворсинок с использованием иглы через брюшную полость или шейку матки.
- Амниоцентез — ещё одно исследование, которое проводится после 15 гестационной недели. Эта процедура включает в себя взятие образца амниотической жидкости путём вставки иглы через живот, управляемой ультразвуковой визуализацией.
Хромосомы в образцах, собранных из обоих диагностических тестов, анализируются для выявления хромосомных аномалий у растущего плода.
Пренатальный скрининг и диагностическое тестирование являются необязательными. Если вы решите отказаться от тестирования, а у вашего ребёнка появятся признаки хромосомной аномалии при родах, специалист выполнит анализ крови у новорождённого, чтобы определить диагноз.
Почему мальчики не болеют
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
Признаки ларингита у детей до года
Дети 4 — 5 лет могут объяснить взрослым, что их тревожит и где больно, но у младенцев сложно определить признаки болезни. Ребёнок не сможет жаловаться на плохое самочувствие.
Важно обратить внимание на физическое состояние и поведение ребёнка. Немедленно обратитесь к специалисту, если наблюдаете следующие проявления ларингита у младенцев:
- кроха серьёзно простудился;
- ребёнок вялый и беспокойный;
- малыш капризный, и его крик сопровождается хрипами и сухим удушливым кашлем;
- в лёгких выслушиваются шумы и свистки;
- носогубный треугольник голубоватого цвета (это один из самых важных сигналов, поскольку синий цвет указывает на прогрессирование болезни).
Когда обнаруживается ларингит у младенцев, лечение назначает только квалифицированный педиатр.
Признаки синдрома Эдвардса
У детей, рождённых с трисомией 18, могут быть некоторые или все из этих характеристик:

- пороки развития почек;
- структурные дефекты сердца при рождении (дефект межжелудочковой и предсердной перегородок, открытый артериальный проток);
- кишечник, выступающий вне тела (омфалоцеле);
- атрезия пищевода (непроходимость пищевода);
- умственная отсталость;
- отставание в развитии;
- дефицит роста;
- трудности с кормлением;
- трудности с дыханием;
- артрогрипоз (тугоподвижность суставов).
Некоторые физические пороки развития, ассоциированные с синдромом Эдвардса:
- небольшая голова (микроцефалия);
- низко расположенные, неправильно сформированные уши;
- аномально маленькая челюсть (микрогнатия);
- расщелина губы / волчья пасть;
- перевёрнутый нос;
- узкие, широко посаженные глаза (глазной гипертелоризм);
- опускание верхних век (птоз);
- короткая грудная кость;
- сжатые руки;
- недоразвитые большие пальцы и / или ногти;
- сращение второго и третьего пальцев ноги;
- косолапость;
- у мальчиков неопущенние яичек.

В утробе наиболее распространённой характеристикой являются аномалии сердца и ЦНС. Наиболее частая внутричерепная патология — наличие кист хориоидного сплетения, которые являются карманами жидкости в мозге. Иногда проявляется избыток амниотической жидкости.
Эпидемиология трисомии 13-й хромосомы
Синдром Патау не очень распространён: только у 1 из 16 000 детей есть это расстройство. 95 % детей с трисомией 13 умирают до рождения.
Средний возраст выживаемости таких детей составляет 2,5 дня, только 1 из 20 малышей выживает дольше полугода. Некоторые дети дорастают до подросткового возраста и, похоже, живут лучше, чем можно было ожидать, исходя из сообщений от тех, кто умирает в перинатальном периоде. Многоуровневое исследование показало, что частота выживаемости до 5 лет для трисомии 13 составляет 9,7 %, причём самые низкие показатели смертности среди девочек. Отчёты о взрослых с синдромом Патау встречаются редко.
Подходы к диагностике гипертермического синдрома у детей

При выявлении ребёнка с лихорадкой важно установить вид лихорадки, тип температурной кривой. Необходимо тщательно собрать жалобы и анамнез
Важно провести полноценный осмотр больного, оценить функцию дыхательной, нервной и сердечно-сосудистой системы. Врач должен попытаться установить связь лихорадки с возможными этиологическими факторами.
Немаловажным является правильно измерять температуру тела. Проводить процедуру необходимо с момента ухудшения состояния больного и периодически её повторять. На приборе устанавливаются минимальные цифры. Подмышечная впадина, куда должен быть установлен ртутный медицинский термометр, должна быть сухой, без воспалительных изменений
Процедура измерения температуры длится 10 минут, при этом важно создать плотный контакт измерительного прибора с кожей
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
От одной боли к другой

Фото: Ефим Эрихман
Через полгода Марине сделали первую операцию – хейлоринопластику. Это пластика верхней губы и носа: у Марины почти не было лица – нос и рот были одной полостью. То, что сделали хирурги, родители считают фантастической работой. У ребенка нет проблем с мимикой, работают мышцы лица. «Выглядит необычно, да, но по сравнению с тем, что было – это просто волшебно. Тогда полость абсолютно не закрывалась», – рассказывает Алана. После этого Марина перенесла еще две операции. Сами по себе они не были тяжелыми – непредсказуема всякий раз реакция ее организма.
Синдром Патау – это прежде всего совокупность нарушений развития. У Марины, помимо проблем, связанных с расщелиной на лице, нет зрения, неправильно расположено сердце, по-своему работает кишечник, случались приступи эпилепсии.
Весь диагноз состоит из мелочей, почти каждая из которых по отдельности вполне совместима с жизнью. Но они вместе, и жизнь ребенка из-за этого непредсказуема.
Марина проходит симптоматическое лечение и реабилитацию. На сеансах массажа ее почти слепили заново – проработали мышцы так, что она смогла сама двигаться. Она даже научилась плавать через два года регулярного посещения бассейна, хотя в основном тренер водит ее по воде.
В три года ей пришлось прекратить все занятия. У Марины случился кризис – начались истерические припадки, пропал сон. Она кричала несколько месяцев. Никто не мог понять, что это. Никто не знал, как это лечить.
«Ее невозможно было вывести из этого состояния, – рассказывает Алана – Она не плакала, а именно кричала – истошно, с рыком, мучением. Она билась головой, вырвала себе все волосы».
Так продолжалось почти полгода. Марина с мамой и няней лежали в Центре паллиативной помощи в Чертаново. Обезболивающие и успокоительные не давали никакого эффекта. Врачи обследовали все: объяснение этому не нашли.
О психологических трудностях
Я не могу сказать, что работа с такими детьми — это особенно тяжелая область медицины с психологической точки зрения. Да, в таких семьях может быть непростая ситуация, в которую ты окунаешься с головой. Но если не отстраняться, не ахать и охать, а вникать, то оказывается, что тяжелая болезнь не означает депрессию и несчастье. Бывают разные ситуации, но в основном очень воодушевляет настрой людей. Вроде бы положение ужасное, а родители чудеснейшие, они делают все, что нужно, стараются. И их жизнь гораздо осмысленнее и часто счастливее, чем у многих других людей, в том числе тех, которые их жалеют.
Причины
23 пары хромосом, унаследованных от родителей, в норме содержит каждая клетка организма человека. Когда соединяются сперматозоид и яйцеклетка, образуя эмбрион, объединяются их хромосомы. Ребёнок получает 23 хромосомы из сперматозоида отца и 23 от яйца матери — всего 46.
Иногда яйцеклетка или сперматозоид имеют неверное число хромосом. Поскольку клетки отца и матери соединяются, ребёнку передаётся это искажение.
«Трисомия» значит, что ребёнок имеет дополнительную хромосому во всех или некоторых клетках. При трисомии 18 у малыша есть три хромосомы 18. Это приводит к ненормальному развитию многих органов у крохи.
Как правило, трисомия 18 вызвана содержанием лишней хромосомы 18 в каждой клетке. Около 5% пострадавших людей имеют дополнительную хромосому 18 в отдельных, а не во всех клетках. Это мозаичная трисомия. Она может быть очень тяжёлой или едва заметной, в зависимости от числа клеток, имеющих лишнюю хромосому.
В редких случаях нет дополнительной хромосомы; часть длинной ветви хромосомы 18 соединяется с другой хромосомой во время образования сперматозоидов и яйцеклетки или в начале развития эмбриона. В этом случае индивидуум имеет 2 хромосомы 18 и ещё добавочный материал из хромосомы 18, который прикреплён к другой хромосоме. Это явление получило название транслокация.
Дополнительный генетический материал вызывает аномалии развития так же, как наличие всей дополнительной хромосомы. Признаки и симптомы этой формы трисомии зависят от количества хромосомного материала, который был перенесён в другую хромосому.
Симптомы синдрома Патау
- тяжелые пороки, которые могут привести к гибели плода;
- пренатальная гипертрофия (недостаток веса ребенка);
- врожденные аномалии головного мозга;
- небольшая окружность головы у новорожденного;
- низкий, скошенный лоб у ребенка, узкие глазные щели, запавшая переносица;
- двусторонние расщелины лица («заячья губа», «волчья пасть»);
- деформация ушных раковин;
- врожденные пороки сердца у ребенка;
- нарушения костно-мышечной системы;
- умственная отсталость;
- нарушение аппетита, отказ от груди;
- многоводие, повышенная раздражительность, плохой сон у матери.
Если Вы обнаружили у себя схожие симптомы, незамедлительно обратитесь к врачу. Легче предупредить болезнь, чем бороться с последствиями.
Причины развития синдрома Патау
Трисомия 13 обычно вызывается погрешностью в делении клеток.
Обычно трисомия 13 не наследуется и является результатом случайных событий при формировании яйцеклеток и сперматозоидов у здоровых родителей. Погрешность в делении клеток приводит к аномальному числу хромосом в яйцеклетке или сперматозоиде. Если один из них вносит вклад в генотип ребёнка, у него будет лишняя хромосома 13 в каждой клетке организма.
Для женщины, у которой никогда не было беременности или ребёнка с хромосомной патологией, риск развития плода с трисомией 13 повышается с возрастом матери. Для женщины, имеющей в прошлом затронутую этой аномалией беременность, или у неё был ребёнок с синдромом, риск рецидива обычно составляет 1 %.
С другой стороны, частичная трисомия 13 может быть унаследована. Незатронутый родитель может иметь перегруппировку генетического материала между хромосомой 13 и другой хромосомой. Эти перегруппировки называются сбалансированными транслокациями, так как не существует лишнего или отсутствующего генетического материала из хромосомы 13. Человек со сбалансированной транслокацией, включающей хромосому 13, имеет увеличенный шанс передать дополнительный материал из неё своим детям.
Людям, заинтересованным в изучении их личного риска иметь ребёнка с хромосомной патологией, следует поговорить со своим врачом или специалистом по генетике. Это поможет родителям:
- детально проанализировать результаты хромосомных тестов;
- определить варианты диагностики для будущей беременности.
О бессмысленной борьбе
В государственной медицине ты не можешь стать борцом: системно исправить ничего нельзя. Один мой знакомый работал начмедом — заместителем главного врача по лечебной работе. В какой-то момент он понял, что получает по разным каналам намного больше, чем рядовые врачи. И он пришел к коллегам по администрации и сказал: «Ребята, зачем нам столько лишних денег? Давайте их перераспределим между врачами». Его тут же выгнали. Другой мой знакомый, еще один добросовестный начмед, тоже пытался что-то изменить. И натыкался на такую же стену, которую строили главный врач и другие его заместители. То есть существует верхушка, которая не даст тебе ничего сделать
Второй по важности врач в больнице не может ничего изменить!
Когда я работал в городской больнице, я постоянно чувствовал, как со всех сторон жмет, все планомерно становится хуже. Сверху (из министерства или департамента) приходят распоряжения, которые не подкрепляются ни ресурсами, ни деньгами. Администрация издает приказы, которые бессмысленны, невыполнимы, а иногда и незаконны. Путаются экономические стандарты и медицинские протоколы. Страшная некомпетентность на уровне управления и совершенное непонимание ни своих работников, ни лечебного процесса. С тех пор, как я ушел, поменялись и министр, и заммэра, и глава департамента. Но лица моих бывших коллег по больнице все печальнее.
Интервью для Medportal.ru 2014 г.
Дарья Саркисян
Фотограф: Максим Копосов
Диагностика синдрома вегетативной дистонии
В диагностике заболевания важное место занимают симптомы, а именно их развитие и течение. На этом фоне особое значение уделяется сбору жалоб и анамнеза
Далее доктор осматривает пациента, осуществляет мониторинг АД, забор фармакологических и физических проб, изучает частоту сокращения сердца, производит тщательную оценку вегетативных индексов. Для постановки на 100% верного диагноза могут понадобиться дополнительные процедуры, такие как кардиоинтервалография или электрокардиография. После вышеупомянутых исследований обычно проводится доплерография сосудов головного мозга, шеи и сердца.





