Методы терапии
Прежде всего, лечение заключается:
- в назначении бета-адреноблокаторов;
- в хирургическом вмешательстве при патологиях клапанов и аорты;
- в хирургической коррекции патологий позвоночника.
Из бета-блокаторов пациентам целесообразно назначать препараты в виде пропранолола или атенолола. Это помогает предотвратить серьёзные осложнения со стороны сосудов и сердца. Бета-блокаторы уменьшают интенсивность сокращения сердечной мышцы, снижая её нагрузку, останавливают процесс расслоения аорты и снижают риск развития аневризмы. Если аневризма достигает критических размеров, пациентам показано хирургическое вмешательство.
В качестве консервативного лечения сколиоза, обычно, применяют фиксацию позвоночника, но если он искривлён от 40 градусов и более, операция является более предпочтительным методом.
Всем пациентам, страдающим синдромом Марфана, нужно каждый год проходить обследование у невролога, кардиолога и окулиста, с генетическим консультированием по показаниям.
Диагностика
К сожалению, на данный момент специфической диагностики нет. Диагноз ставится на основании жалоб и эпизодов лихорадки.
В общем анализе крови могут быть незначительные изменения – увеличение СОЭ, нейтропения (уменьшение количества нейтрофилов). Посевы на микрофлору из горла часто не дают никаких результатов при данном заболевании.
PFAPA синдром — это диагноз исключения. В первую очередь исключаются другие причины рецидивирующей лихорадки: инфекция, воспалительные заболевания кишечника, лихорадка при лимфоме Ходжкина, циклическая нейтропения и другие.
Важными диагностическими критериями являются:
- Более 3 эпизодов лихорадки продолжительностью до 5 дней и возникающих регулярно на фоне полного здоровья;
- Фарингит и лимфаденопатия или афтозные язвы;
- Хорошее здоровье между эпизодами и нормальный рост;
- Разрешение симптомов в течение нескольких часов после лечения преднизолоном в виде однократной дозы или двух доз с интервалом от 12 до 48 часов.
Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов
Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков
Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Болезни радужной оболочки
Синдрому Марфана нередко сопутствуют изменения в радужной оболочке — это связано с повышенной растяжимостью тканей. Могут возникнуть колобомы, гипоплазия или атрофия радужки с нарушением ее диафрагмальной функции. Колобома — это дефект, проявляющийся в отсутствии части глазной оболочки. Обычно имеет грушевидную форму и располагается в нижней части радужки. К наиболее ранним проявлениям у людей с синдромом Марфана относят гипоплазию стромы радужки, особенно ее пигментной зрачковой каймы. Слабость дилататора (мышцы-расширителя) у больных не позволяет достичь полного расширения зрачка даже с помощью мидриатиков.
Лечение синдрома Марфана
К сожалению, на сегодняшний день лекарственные методы терапии этой генетической патологии еще не разработаны
Однако пациентам с синдромом Марфана важно соблюдать все назначения врачей, чтобы устранить симптомы патологии и замедлить темпы ее развития
Лечение зависит от клинических проявлений болезни:
- при аневризме аорты назначают препараты, которые снижают частоту и силу сердечных сокращений, снимая избыточную нагрузку на сосуды;
- пациентам с синдромом Марфана часто назначают антигипертензивные препараты для снижения артериального давления;
- хондроитин и глюкозамин относятся к естественным компонентам соединительной ткани — их прием улучшает структуру хрящей и предупреждает патологии суставов;
- для стимуляции образования коллагена выписывают специальные БАДы — L-карнитин, витамины из групп С, D, Е, В, а также кальций, цинк и другие пищевые добавки.
Пациентам противопоказаны физические нагрузки, постоянная активность, травмоопасные игры. Рацион питания людей с синдромом Марфана должен быть насыщен белками, полезными жирными кислотами, микро- и макроэлементами. Для поддержки структур скелета пациентам с мутацией в гене FBN1 показано ношение корсетов, укрепление мышц с помощью ЛФК и оздоровительного массажа.
В некоторых случаях может помочь только хирургическое лечение — операции по замене части аорты, клапанов, исправлению костных патологий или коррекции патологий глаза, которые существенно снижают риски опасных осложнений.
Катаракта и глаукома при синдроме Марфана
Осложнения при эктопии хрусталика могут проявиться в его помутнении — тогда развивается катаракта. При этом качество зрения постепенно падает, и процесс необратим. Лечить заболевание надо своевременно. Обычно операция назначается при помутнении примерно 50% хрусталика. Современные методы в офтальмологии позволяют без труда справиться с этим нарушением зрения. Для этого проводится хирургическая операция: хрусталик удаляется посредством факоэмульсификации, а вместо него имплантируется интраокулярная линза. Она заменяет естественную и обеспечивает высокое качество зрения надолго.
В данной ситуации глаукома лечится теми же способами, что и обычно. Назначаются средства, понижающие давление и увеличивающие отток внутриглазной жидкости. В более тяжелых случаях может быть назначена хирургическая операция, направленная на улучшение оттока жидкости.

Может ли слабость соединительной ткани быть опасной?
В связи с нарушением клапанной функции существует повышенный риск развития сердечно-сосудистой патологии. В период беременности у женщин, имеющих такие изменения, увеличивается вероятность расслоения аорты, из-за гиперсекреции эстрогенов, замедляющих поступление коллагена и эластина в аорту.
Слабость связочного аппарата глаза у взрослых проявляется глаукомой или формированием дегенерации сетчатки с последующей ее отслойкой. В период активного быстрого роста наблюдается развитие тяжелых сколиозов и протрузии вертлужной впадины. Рентгенологически эти признаки хорошо выявляются на снимках.
ОНЛАЙН-ЗАПИСЬ в клинику ДИАНА
Вы можете записаться по бесплатному номеру телефона 8-800-707-15-60 или заполнить контактную форму. В этом случае мы свяжемся с вами сами.
Симптоматика со стороны разных органов и систем
Со стороны сердца и сосудов у больных чаще всего выявляют пролапс клапана и аортальную аневризму. Если корень аорты и её восходящая часть патологически изменены, это чревато серьёзными осложнениями. Как правило, повреждение интимы аорты происходит там, где нагрузка на неё особенно велика. Происходит постепенное расширение сосуда либо коронарный синус может внезапно разорваться.
Расширение корня аорты диагностируют у половины пациентов в детском возрасте и примерно в 60-80% случаев у взрослых. Аортальную недостаточность выявляют с помощью аускультации, в виде диастолического шума, прослушиваемого над клапаном. При растяжении клапанных створок и хорд может возникнуть пролапс митрального клапана с систолическим щелчком и шумом разной интенсивности. Не исключена возможность развития эндокардитов бактериальной природы как на аортальном, так и на митральном клапане.
Со стороны опорно-двигательной системы важным признаком является высокий рост пациента, с размахом рук, значительно превышающим их высоту. При более тщательном осмотре можно выявить арахнодактилию большого пальца с выступлением его дистальной фаланги за край руки, сжатой в кулак. Грудная клетка имеет форму киля со смещением наружу либо воронки — со смещением внутрь. Суставы часто гиперподвижны, с незначительными контрактурами в области локтей. Колени могут быть выгнуты назад, нередко плоскостопие и искривление позвоночника по типу кифоза и сколиоза. Также наблюдается слабое развитие подкожно-жировой клетчатки.
Эктопия хрусталика (его подвывих либо он смещён вверх) является основным признаком синдрома Марфана со стороны зрительной системы. Также типичны явления иридоденеза, когда радужная оболочка глаз дрожит. Если поражён хрусталик, край его дислоцирован, что можно увидеть, когда зрачки пациента не расширены. Близорукость со спонтанным отслоением глазной сетчатки тоже нередки.
Органы дыхания часто дают патологическую симптоматику в виде кистозной лёгочной болезни и спонтанного пневмоторакса, склонного к постоянному рецидивированию. В результате пациентов часто беспокоят одышка и боли в грудной клетке.
Что касается симптомов со стороны нервной системы, обследование показывает, что дуральный мешок, который окружает спинной мозг, сильно расширен в области поясницы и крестца. Это вызывает боли как в голове, так и в спине, а если, при этом, имеется неврологическая патология, функции мочеиспускания и дефекации могут быть значительно нарушены.
Online-консультации врачей
| Консультация неонатолога |
| Консультация инфекциониста |
| Консультация эндокринолога |
| Консультация пульмонолога |
| Консультация офтальмолога (окулиста) |
| Консультация психолога |
| Консультация хирурга |
| Консультация гомеопата |
| Консультация кардиолога |
| Консультация нейрохирурга |
| Консультация гинеколога |
| Консультация нарколога |
| Консультация генетика |
| Консультация общих вопросов |
| Консультация массажиста |
Новости медицины
6 простых привычек, чтобы круглый год не болеть простудами: рекомендуют все врачи,
17.03.2021
Морепродукты становятся вредными для здоровья?,
05.01.2021
Digital Pharma Day. Будьте во главе digital-трансформации фармацевтической индустрии,
09.10.2020
В сети EpiLaser самые низкие цены на ЭЛОС эпиляцию в Киеве,
14.09.2020
Новости здравоохранения
Эксперт назвала три отличия простуды от COVID-19,
05.01.2021
В мире более 86 миллионов случаев COVID-19,
05.01.2021
Скорость распространения COVID-19 зависит от климатических условий,
11.06.2020
Исследователи насчитали шесть разновидностей коронавируса,
11.06.2020
Советы родителям ребенка с синдромом Марфана
Конечно, грустно и горько осознавать, что у ребенка имеется такое опасное заболевание. Однако только правильное поведение родителей поможет преодолеть трудности, в том числе создать нужный психологический настрой. Нужно научиться жить с этой проблемой и постоянно держать под контролем состояние ребенка
Важно найти хороших специалистов, которые будут заниматься его здоровьем
Следует также научить его относиться с достоинством к своему состоянию, не реагировать на возможные насмешки со стороны. Лучше привить ему любовь к занятиям, которыми он сможет заниматься в дальнейшем: пусть это будет программирование или, например, музыка. Если диагноз был поставлен уже в старшем возрасте, то придется отказаться от некоторых видов спорта.
Важно также наладить хороший контакт с учителями, объяснив им, что это заболевание предполагает некоторые особенности. Например, сидеть ему лучше за первой партой из-за проблем со зрением, не стоит испытывать слишком сильных физических нагрузок
Каждому родителю хочется обеспечить своему ребенку счастливое детство. Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны.
Симптомы Болезни (синдрома) Марфана:
Клинические симптомы заболевания разделяют на несколько групп, которые отражают точную локализацию проявлений соединительнотканной (мезенхимальной) дисплазии:
- костно-суставные расстройства (астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий). Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей (малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие);
- изменения внутренних органов (аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана, гипоплазированное, расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком длинный и гипопластичный кишечник);
- нарушения в системе зрения (голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома);
- расстройства центральной нервной системы (анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства);
- Расстройства гипофизарно-адреналовой системы (высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства);
- нарушения сердечно-сосудистой системы (нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность, пролапс митрального клапана, нарушения внутрисердечной гемодинамики, расширение корня аорты, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования). У больных наблюдается мышечная слабость и снижение активности при физических нагрузках.
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Почему при определении признаков синдрома Марфана нужно обратиться к врачу?
Сама по себе генетическая аномалия совместима с жизнью. Однако опасны последствия болезни, вызванной FBN1 мутацией:
- разрывы крупных сосудов, чаще всего — аорты;
- хроническая сердечная недостаточность — неспособность сердца обеспечивать необходимую работу для кровоснабжения всех органов;
- снижение остроты зрения или полная потеря зрительной функции.
Разрыв аневризмы аорты или другого магистрального сосуда часто заканчивается моментальным летальным исходом. Хроническая сердечная недостаточность может перейти в острую форму, а без экстренной медицинской помощи также привести к фатальным последствиям — внезапной коронарной смерти. Именно эти осложнения чаще всего приводит к гибели детей с синдромом Марфана. Особая опасность ждет женщину с синдромом мутации гена FBN1 во время беременности: повышенная нагрузка на аорту в разы увеличивает риск ее разрыва.
Чтобы предупредить развитие опасных осложнений и компенсировать возникающие нарушения, родителям нужно как можно раньше обратиться за медицинской помощью при первом подозрении на синдром Марфана у ребенка
При этом важно не только однократно провести обследование, но и стать на учет к врачам, которые занимаются коррекцией проявлений синдрома:
- специалисту по генетическим болезням;
- кардиологу;
- ортопеду-вертебрологу;
- дерматологу;
- офтальмологу;
- гастроэнтерологу.
Список специалистов зависит от степени выраженности заболевания, при этом регулярно необходимо проходить комплексные профилактические осмотры для раннего выявления новых нарушений.
Синдром Марфана — болезнь гениев?
С синдромом Марфана связаны не только многочисленные поводы для обращения к врачам. Часто люди с мутацией гена FBN1 компенсируют физические проявления болезни интеллектуальными способностями, поэтому это генетическое заболевание даже называют «синдромом гениев». Считается, что повышенный выброс адреналина из-за патологических изменений в надпочечниках определяет высокий тонус умственной и психической активности у таких пациентов. Именно поэтому в числе людей с синдромом Марфана можно найти известных личностей. Например, Юлию Цезарю, Аврааму Линкольну и Шарлю де Голлю патология не помешала стать известными политическими деятелями; Ганс Христиан Андерсен и Корней Чуковский создали уникальные литературные произведения, а Никколо Паганини прославился как гениальный музыкант.
Современные знаменитости также не скрывают свои недостатки и становятся еще более популярными из-за генетического дефекта. Например, солисту американской рок-группы Deerhunter Брэдфорду Коксу нетипичная внешность придает особый шарм, а испанский актер Хавьер Ботет очень востребован, поскольку правдоподобно и талантливо играет отрицательных героев в голливудских фильмах ужасов (рис. 6).
К каким докторам следует обращаться если у Вас Болезнь (синдром) Марфана:
Кардиолог
Офтальмолог
Кардиохирург
Ортопед
Генетик
Терапевт
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни (синдрома) Марфана, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Причины синдрома Марфана
Данное генетическое заболевание вызвано дефектом гена FBN1 в длинном плече 15 хромосомы. Этот ген кодирует белок гликопротеин фибриллин-1, который отвечает за прочность и эластичность соединительной ткани. Соответственно, все проявления патологии связаны с тем, что соединительнотканные структуры в организме человека теряют свои нормальные свойства.
Наследуется мутация по аутосомно-доминантному признаку, то есть дети получают патологический ген от родителей, которые страдают от патологии. При этом шанс ребенка получить мутацию от одного из родителей составляет 50% (рис. 1). Синдром не передается через поколение: здоровые дети больных родителей не могут передать ген своим потомкам.
Однако примерно у 25% людей с синдромом Марфана никто из родителей не оказывается носителем аномалии гена FBN1: в таком случае мутация развивается спонтанно.
До сих пор не выявлено определенных факторов риска развития этого генетического нарушения: заболевание встречается одинаково часто среди мужчин и женщин, а его распространенность не зависит от расы или этнической группы. Частота заболеваемости у этой патологии составляет примерно 1 случай на .
Если клинические признаки мутации ярко выражены, заподозрить болезнь можно уже в первые месяцы жизни ребенка, но стертые формы заболевания часто проявляются уже во взрослом возрасте, когда пациент обращается к врачам по поводу различных проявлений синдрома.
Важно! Не стоит записываться на генетическое обследование в качестве медосмотра. Поиски «поломки» гена FBN1 оправданы только в случае, если болезнь проявляет себя характерными признаками: бессимптомное носительство этой мутации невозможно
Если у одного из родителей установлен этот диагноз, будущей маме следует пройти генетическое обследование еще до родов. Это позволит заранее узнать, передалась ли аномалия ребенку.
Патологии сетчатки при синдроме Марфана
Из-за слабости соединительной ткани подвержена растяжению также сетчатка глаза. В результате этого повышается риск появления периферических хориоретинальных дистрофий — локальных истончений сетчатки, которые могут спровоцировать отторжение слоя светочувствительных клеток от пигментного эпителия. Такое нарушение очень опасно: начинает падать острота зрения, ухудшается восприятие света и цвета.
Признаками отслойки сетчатки могут быть следующие проявления:
- вспышки, искры в глазах — такое явление называется фотопсией;
- искажение формы, размера и оттенка объектов — метаморфопсия;
- «мушки» и черные точки перед глазами как следствие повреждения ретинального сосуда;
- выпадение из поля зрения отдельных элементов в видимой картинке — это признак того, что отслоение началось в центральной зоне сетчатки;
- появление темной пелены, охватывающей все большую область, снижение периферийной видимости.
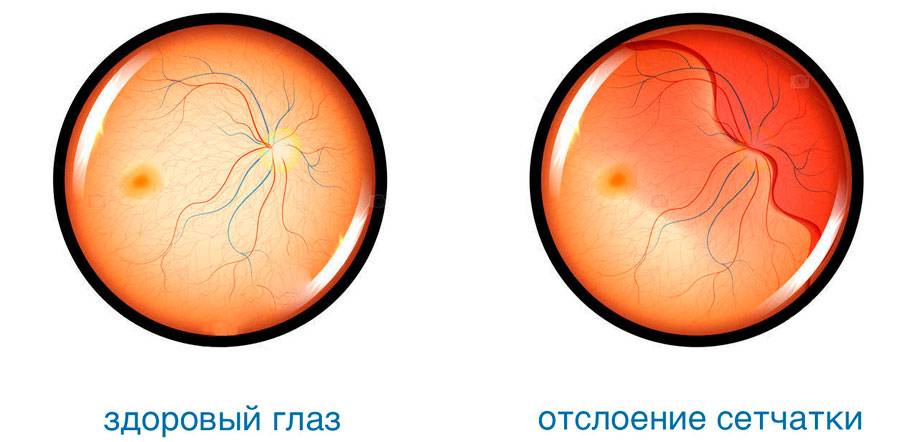
Отслоение сетчатки сегодня поддается успешному лечению различными методами. Наиболее эффективным из них является лазерная коагуляция — прижигание поврежденных участков с целью надежного их соединения с сосудистой оболочкой.







Очень важная тема! Спасибо, что подняли её. Чтобы не пропустить синдром Марфана у ребёнка, нужно внимательно следить за его ростом, строением тела и состоянием сердца, а при любых сомнениях сразу обращаться к врачу. Ранняя диагностика помогает вовремя начать лечение и поддержать здоровье малыша.