Введение прикорма и дополнительных продуктов питания
После трех месяцев рацион питания начинают расширять за счет использования соков (фруктовых и ягодных), назначая их с 3–5 капель, с постепенным увеличением объема до 30–50 мл, а к концу первого года жизни — до 100 мл. Основные соки: яблочный, грушевый, сливовый и т. д. Фруктовые пюре назначают, увеличивая их количество в рационе питания аналогично с таковым вводимого сока.
В период с 4–4,5 месяцев в рацион питания вводят первый прикорм в виде овощного пюре, приготовленного самостоятельно (или плодоовощных консервов для питания детей грудного возраста — последние без добавления молока).
Далее последовательно назначается 2-й прикорм — каша (10%) из молотого саго или безбелковой крупки. Могут быть использованы безмолочные каши промышленного производства на основе кукурузной и/или рисовой муки, содержащие не более 1 г белка в 100 мл готового к употреблению продукта.
После 6 месяцев в питание можно ввести кисели и/или муссы (безбелковые), которые готовятся с использованием амилопектинового набухающего крахмала и фруктового сока, безбелковый напиток с молочным вкусом Нутриген или низкобелковый молочный напиток PKU «Лопрофин».
С 7 месяцев ребенок с ФКУ может получать низкобелковые изделия , например, спиральки, спагетти, рис или безбелковую вермишель, а с 8 месяцев — специальный безбелковый хлеб.
Психомоторное и умственное развитие
Дефекты в развитии детей с таким синдромом появляются по нескольким причинам:
- снижение зрения и слуха, из-за чего ребенок не в состоянии воспринимать информацию. Вследствие этого происходят и когнитивные нарушения;
- гипотонус мышц — малыши поздно начинают держать голову, переворачиваться, ходить. Страдает моторика;
- проблемы с опорно-двигательным аппаратом;
- деформация неба, зубов и языка вызывает речевые затруднения, поэтому речь таких людей может быть непонятной.
При мозаичной форме заболевания таких дефектов может и не быть, поэтому эти детки достаточно хорошо развиваются, их умственное развитие практически не страдает. Для них разрабатывают специальные программы, и здесь большая роль принадлежит родителям. От того, насколько качественно и стабильно они будут заниматься со своим ребенком, зависят его дальнейшие способности и успехи.
Дети с мозаицизмом очень способные к обучению. Они могут посещать обычный садик и школу, и даже преуспевать больше, чем некоторые их здоровые сверстники.
Их возможности, способности и развитие в целом очень разнятся. На форумах мамочки таких детей делятся своими достижениями и по-разному описывают своих малышей с подобным синдромом.
К примеру, одна мама говорит о том, что ее сын, которому уже 8 лет и который страдает мозаичной формой СД, до сих пор не разговаривает. Хотя они проходят реабилитацию в соответствующем центре.
Тогда как другая мама, у которой младший малыш имеет такое заболевание, утверждает, что он ничем не отличается от старшего, даже более находчивый.
Многие дети с мозаичным типом СД сублимируют свои дефектные особенности в невероятные достижения.
Айя Ивамото — девушка с мозаицизмом из Японии, родители которой рассказали о её болезни только на втором курсе института. Она посещала садик, успешно окончила школу и университет. Изучила два языка: английский и французский, и занимается иностранными переводами. Преподает в школах и вузах, принимала участие в международной конференции.
Совместно с мамой издала книгу о путешествии во Францию. Регулярно поддерживает и борется за права людей с синдромом Дауна.
Раймонд Ху – парень с мозаичным синдромом Дауна из США, который рисует картины с помощью старинной китайской техники. Он создает их акварельными красками и тушью на рисовом бумажном полотне.
В группу людей с подобным дефектом входит много актеров. К одной из наиболее известных таких личностей относится актер Пабло Пинеда. Ему досталась главная роль в фильме «Я тоже». Он также является частым гостем в различных телешоу, где освещает проблемы педагогики и развития детей.
Джейми Бауэр, сыгравший в «Американской истории ужасов», Паскаль Дюкенн, Крис Берк — все эти актеры имеют мозаичный синдром Дауна.
Люди с таким диагнозом находят себя в разных сферах жизни. Тим Харрис – владелец ресторана. Рональд Дженкинс – известный музыкант, приручивший синтезатор. Ему нет равных в электронной музыке. Мигель Томасин — популярный барабанщик, участник группы Reynols. Карен Гафнии – девушка, занимающаяся плаванием. Стала первой, кто преодолел дистанцию в 15 км. Вода при этом достигала температуры +15.
Глядя на такие примеры, сложно предугадать, как будет развиваться ребенок с мозаичной формой синдрома Дауна. Ясно одно: чтобы он стал успешным и развитым, нужна кропотливая работа как со стороны ребенка, так и его родителей.
Фенилкетонурия и интеллект ребенка
Без своевременного начала лечения уровень психического развития может варьироваться от нормального до классической клинической картины фенилпировироградной олигофрении с выраженной умственной отсталостью. Все зависит от остаточной активности фермента, расщепляющего фенилаланин, и предсказать вариант развития болезни заранее пока невозможно.
При своевременной терапии пациенты со всеми формами заболевания достигают норм возрастного развития, психических и когнитивных функций, хотя по недавним исследованиям их IQ несколько ниже, чем у сверстников в среднем. Также могут присутствовать мелкие неврологические проявления: трудность с концентрацией внимания, замедленная реакция и т. д., что может влиять на способность к обучению.
Общие правила улучшения соблюдения диеты при ФКУ
— Регулярный 3-4х разовый в сутки приём аминокислотной смеси без фенилаланина
— Возможность выбора „разрешённых” продуктов
— Включение ребенка в помощь при подготовке еды — соответсвенно возрасту и возможностям
— Блюда похожи, кушать вместе, за одним столом
— Не критиковать вкус, запах, вид аминокислотной смеси
— Нельзя кушать перед ребёнком „запрещённых” продуктов, не имея для него малобелковых продуктов
— Быть последовательным и решительным
— Нельзя разрешать пробовать „запрещённых продуктов” – „легче отказяться от этого, чего не попробовал”
— Постоянный конакт с лечащим врачём, следование его указаниям
Как принимать формулу
— Ежедневно!
— Минимум 3 – 4 дозы равномерно распределенные в сутки
— Лучше всего подготовленный сразу перед использованием
— Тщательно перемешать перед употреблением (тирозин-аминокислота нерастворимая в воде, оседает на дне стакана-> дефицит тирозина)
Лечение фенилкетонурии
ФКУ является единственным наследуемым заболеванием, которое при своевременной диагностике, поддается полному контролю: ведь дети появляются на свет совершенно здоровыми. При детекции заболевания в первые дни жизни с помощью специальной диеты можно предотвратить пагубное влияние фенилаланина на ЦНС. Из рациона больных полностью изымают мясопродукты, рыбу, весь спектр молочных продуктов, яйца, бобовые, грибы. Отдают предпочтение овощам, фруктам, сокам, амилофенам. Диета считается адекватной, если уровень фенилаланина крови находится в пределах 120–240 мкмоль/л. С течением времени проводят расширение диеты. Для компенсации недостатка аминокислот при ФКУ применяют белковые гидролизаты, смеси L-аминокислот, лишенные фенилаланина, но содержащие другие незаменимые аминокислоты. Кроме этого назначаются витамины, минеральные соединения, микроэлементы, препараты железа, ноотропы. Широко применяется физиотерапия. Женщины репродуктивного возраста, получавшие с детства адекватную дието- и медикаментозную терапию, способны воспроизвести здоровое потомство.








Основные лекарственные препараты
Имеются противопоказания. Необходима консультация специалиста.
- Нофелан С (энзиматический белковый гидролизат из белков коровьего молока). Режим дозирования: применяют внутрь в 2–3 порциях в дозах: для детей от 1 года до 2 лет 56–62 г/сут., 2–3 лет — 62–75 г/сут., 3–5 лет — 75–81 г/сут., 5–8 лет — 81–94 г/сут.
- Тетрафен 30 (сухая питательная смесь для питания детей с фенилкетонурией старше 1 года). Режим дозирования: смесь разбавляют водой или соком и употребляют внутрь. Количество смеси на суточный прием определяется строго индивидуально.
- L-карнитин (Элькар) — препарат, улучшающий метаболизм и энергообеспечение тканей. Режим дозирования: принимают внутрь за 30 мин. до еды в средней суточной дозе 10–20 мг/кг/сут. в 2-3 приема в течение 1–2 мес. В год 3–4 курса.
Профилактика Фенилкетонурии (ФКУ) у детей:
Чтобы организовать раннюю диетотерапию и избежать тяжелых церебральных повреждений, нужно проводить массовые скрининги на фенилкетонурию в неонатальном периоде. Это позволяет также избежать нарушения функционирования печени ребенка. Чтобы оценить риск рождения ребенка с рассматриваемым диагнозом, нужно предварительное генетическое консультирование для пар, у которых уже есть ребенок с фенилкетонурией (ФКУ) или у которых есть родственники с такой болезнью.
Женщины с фенилкетонурией до момента зачатия должны строго придерживаться диеты и продолжать ее, пока будут беременными. Это позволит избежать нарушений развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Дети с ФКУ должны наблюдаться участковым педиатром и психоневрологом.
Что такое Фенилкетонурия (ФКУ) у детей —
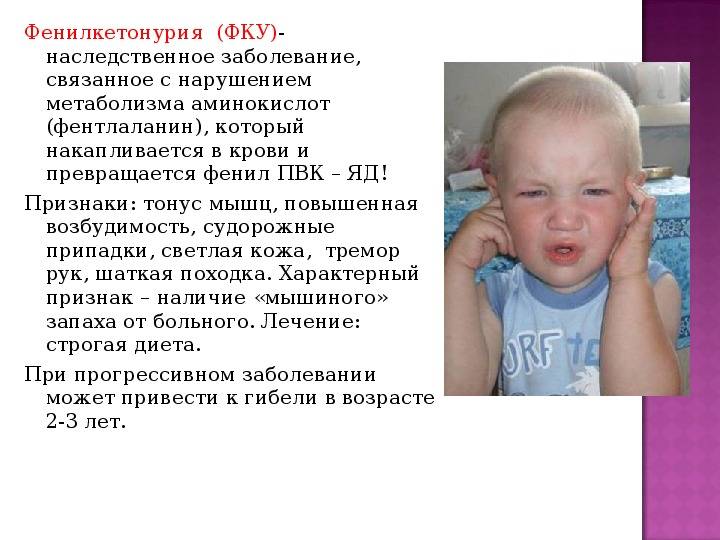
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
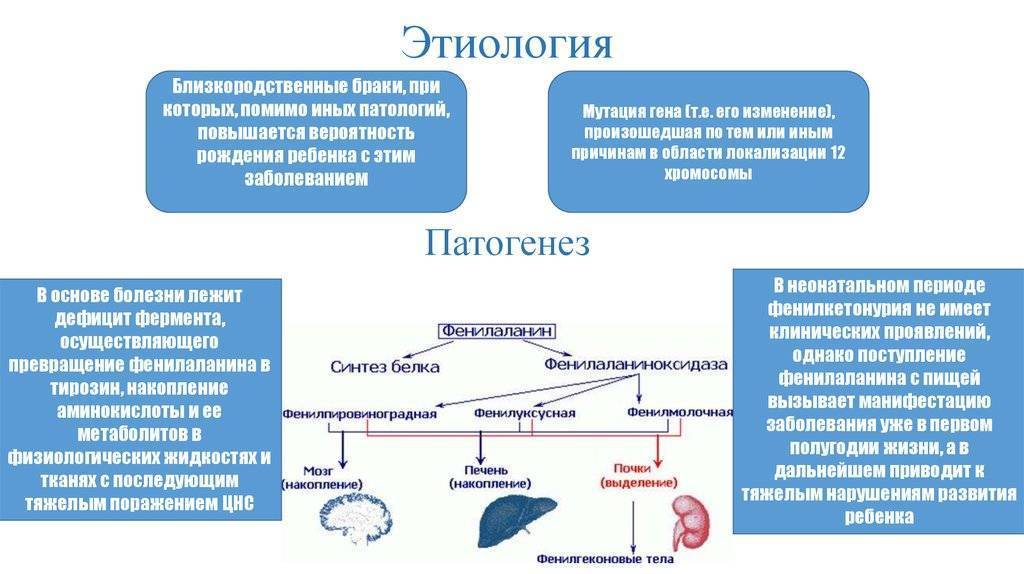
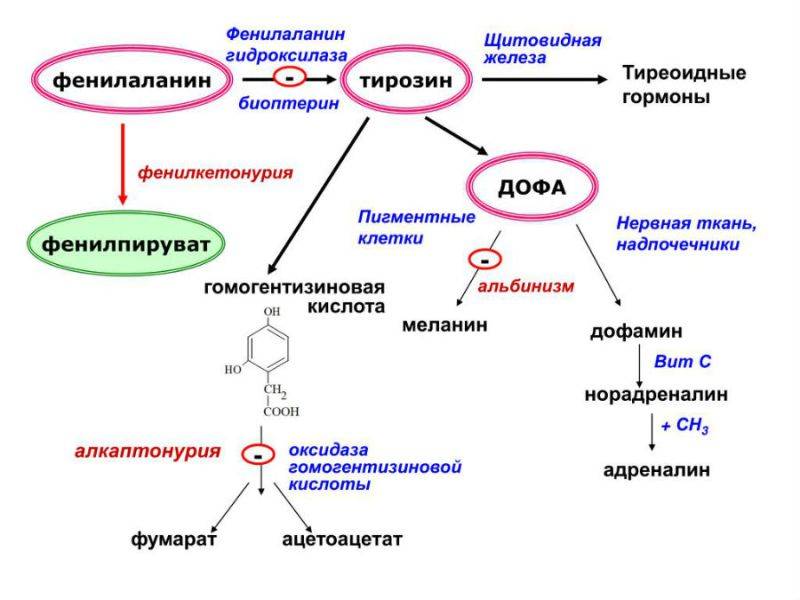
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
|
Название формы |
Уровень фенилаланина |
|
классическая |
выше 20 мг% (1200 мкмоль/л) |
|
средняя |
10,1–20 мг% (600–1200 мкмоль/л) |
|
легкая |
до 8 мг% (480 мкмоль/л) |
Лечение
Основным способом лечения сегодня является своевременное начало диеты. Необходимо ограничить прием пищевых продуктов, в которых содержится фенилаланин, в частности белковых продуктов. Их заменяют белковыми гидролизатами и аминокислотными смесями. В рационе должны преобладать овощи и фрукты, углеводы, животные и растительные жиры.
Строгая диета соблюдается на протяжении 5 лет. Если все меры соблюдены правильно, то к 12–14 годам ребенок может перейти к обычному питанию. Медикаментозное лечение является периодическим и направлено, прежде всего, на устранение судорог и повышение интеллектуальной деятельности ребенка. Кроме того, дети с ФКУ обязательно проходят курс массажа и лечебной физкультуры, а также посещают специальные занятия для развития логического мышления.










Контролировать содержание аминокислоты в организме необходимо и по окончании диеты. Ее должно быть достаточно для нормального развития и роста ребенка, но при этом она не должна накапливаться в тканях.
Статистика фенилкетонурии в Польше
В Поликлинике Метаболических Заболеваний ИМиР (Польша) наблюдается
-
* В общем – ok. 780 больных
-
— мягкая гиперфенилаланинемия – 172 человек
-
— классическая фенилаланинемия — 595 человек
-
— злокачественная гиперфенилаланинемия —
-
дефицит BH4 – 12 человек
Дети с ГФА, рожденные в 2010 году:
-
В общем – 26 новорождённых
-
(полная диагностика: общие анализы крови, фен/тир; двугидроптериновая редуктаза DHPR, тест нагрузки BH4, профиль выделенных из мочи биоптеринов)
-
— 10 детей – классичеслая ФКУ
-
— 1 ребёно — мягкая ФКУ
-
— 12 детей – мягкая ХФА
-
— 1 ребёнок – дефицит PTPS
-
— 2 детей — переходная гиперфенилаланинемия
«Потерянные» случаи — причины
-
Рождённые дома
-
Ранняя выписка из больницы
-
Ошибка в подписи пробы (один код на пробе, другой код приклеен в детской книжке)
-
Забыли взять пробу крови
-
Потерия пробы крови по пути в лабораторию
-
Проба крови взята от одного ребёнка на несколько бумаг
Рекомендации
-
Необходимость проведения скрининга всей популяции новорожденных.
-
Проведение диагностики разными методами в каждом случае гиперфенилаланинемии.
-
Начало диеты с низким содержанием фенилаланина как можно раньше, оптимально до исполнения 21 суток жизни ребенка.
-
Начало лечения сразу при уровнях фенилаланина > 10 мг %, или после нескольких дней ежедневного контроля, если концентрация фенилаланина удерживается в пределах 7-10 мг %
-
Проведение лечения на основе современных препаратов без фенилаланина согласно определенным схемам, с учетом в каждом случае индивидуального потребления фенилаланина.
-
Регулярный мониторинг избранных биохимических показателей в крови больного (например, концентрации фенилаланина, тирозина, цельного белка) а также психического и физического развития больных.
-
Лечение девушек и женщин до зачатия, а также в течение всего периода зачатия, из-за вредного действия высокой концентрации фенилаланина на развитие плода.
-
Продолжение лечения и мониторинг лечения больных взрослых.
Недостаток однозначных лечебных рекомендаций для взрослых
-
„Исчезание” пациентов после окончания 18 года жизни
-
Проблема материнской фенилкетонурии
-
Проблема остеопороза при длительной диете
-
Низкий уровень акцептации („compliance”) у подростков и взрослых обусловлен чрезвычайной обременительностью лечения
-
(правильно лечится меньше, чем 20% взрослых больных)
Что такое Фенилкетонурия (ФКУ) у детей —
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.










Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
|
Название формы |
Уровень фенилаланина |
|
классическая |
выше 20 мг% (1200 мкмоль/л) |
|
средняя |
10,1–20 мг% (600–1200 мкмоль/л) |
|
легкая |
до 8 мг% (480 мкмоль/л) |
Диагноз
Диагноз ставят в первые месяцы жизни на основании данных лаб. исследования. Для выявления фенилкетонурии у новорожденных используют тест Гатри (см. Гатри метод). При обнаружении повышенной концентрации фенилаланина в крови (более 4 мг: 100 мл) применяют более точные количественные методы определения содержания этой аминокислоты — энзиматический метод или флюориметрию (см.). Содержание фенилаланина в крови при фенилкетонурии не менее 15 мг/100 мл. В моче обнаруживают фенилиировиноградную, фенилуксусную, фенилмолочную кислоты. При исследовании мочи используют треххлористое железо, при добавлении которого моча приобретает грязнозеленую окраску, или 2,4-динитрофенил гидразин, при взаимодействии с к-рым моча мутнеет и становится ярко-желтой. Для исследования мочи применяют также индикаторные
бумажки (фенистикс, биофан), пропитанные треххлористым железом.
Фенилкетонурию следует дифференцировать с гиперфенилаланинемией, нередко наблюдаемой у недоношенных детей в связи с недостаточностью фермента фенилаланин-4-гидроксилазы. При гиперфенилаланинемии содержание фенилаланина в крови составляет менее 15 мг/ 100 мл. Гиперфенилаланинемия обычно исчезает в течение 1—3 месяцев., иногда после назначения аскорбиновой кислоты.
Симптомы Фенилкетонурии (ФКУ) у детей:
Новорожденный ребенок не похож на больного. Симптомы фенилкетонурии (ФКУ) начинают быть заметны в возрасте 2-6 месяцев. Типичные проявления:
- отсутствие интереса к окружающему миру
- выраженная вялость
- рвота
- беспокойство
- повышенная раздражительность
С 6 месяцев у малыше заметно отставание в психическом развитии. У меньшинства детей это олигофрения в слабой степени. А более чем у половины детей фиксируют идитию. Рост малыша с ФКУ может быть нормальным или сниженным. Зубки режутся поздно, череп может иметь размеры меньше нормы. Сидеть и ходить ребенок с фенилкетонурией начинает поздно.
Детей с рассматриваемым диагнозом можно отличить по позе и походке. Они широко расставляют ноги, сгибая их в тазобедренном и коленных суставах. Шаги мелкие. При ходьбе ребенок покачивается. Сидят они в так называемом положении портного – поджав ноги, поскольку у них повышен мышечный тонус.
При фенилкетонурии (ФКУ) дети обычно имеют голубой цвет глаз и светлый оттенок волос. Кожа почти не пигментирована. От ребенка слышен «мышиный» запах. В некоторых случаях у больного могут быть припадки эпилепсии, но они проходят по мере взросления ребенка.
Другие типичные симптомы ФКУ у детей:
- дермографизм
- потливость
- повышенная чувствительность к солнечным лучам и травмам
- акроцианоз
- тяжёлая экзема
- дерматит
- склонность к запорам
- артериальная гипотония
- расстройства аутистического спектра
- гиперактивность
Если не провести вовремя лечение, уровень интеллекта ребенка будет составлять менее 50. В возрасте 18 месяцев могут появиться судорожные приступы. Исчезают они спонтанно. В раннем возрасте приступы часто проходят в форме инфантильных спазмов, далее становятся тоникоклоническими припадками.








Когда появляются первые симптомы патологии у детей?
Опасность фенилкетонурии — в почти полном отсутствии выраженных симптомов болезни до этапа выраженного повреждения головного мозга. В первые месяцы после рождения ребенок кажется здоровым, в некоторых случаях накопление фенилаланина проявляется атипичным запахом пота, мочи, кожной экземой, которую часто принимают за явления атопического дерматита.
Как правило, первая яркая симптоматика связана с отклонениями от возрастных норм в моторных навыках — умении садиться, вставать, разговаривать. Наиболее заметными признаки поражения ЦНС становятся на втором году жизни: появляются стереотипии, повторяющиеся движения руками, головой, повышенная раздражительность, сенситивность, что вместе с нарушением речевого и моторного развития нередко принимают за симптомы расстройства аутичного спектра.
Терапия фенилкетонурии: диета как основа лечения
Основа терапии — строгая диета с ограничением белка и дополнением специальных протеиновых продуктов, подходящих для больных с нарушением выработки фермента, и подходящими витаминными комплексами. Хотя рацион питания достаточно скудный, врачи настойчиво рекомендуют переводить на него ребенка сразу же после установления диагноза и придерживаться диеты пожизненно.
Отказ от диеты во взрослом возрасте после окончания формирования отделов головного мозга и связей между ними вызывает нервно-психические расстройства и небольшую инволюцию когнитивных навыков.
Есть мнение, что диетотерапию стоит начинать применять у детей по прошествии нескольких месяцев, чтобы создать депо нутриентов и насытить организм на первом этапе полезными белками.
Хотя первые месяцы жизни у ребенка с фенилкетонурией, как правило, не сопровождаются выраженной симптоматикой, такое решение сегодня признано ошибочным и вредным. За месяцы без диеты уровень фенилаланина успевает достигнуть концентрации, сказывающейся на состоянии ЦНС. На особое питание ребенка надо переводить в первые же дни.
Из рациона питания больных исключают или ограничивают высокобелковые продукты: мясо, рыбу, яйца, сыр, орехи, сою. Потребность в протеине восполняется за счет заменителей на основе L-аминокислот. Диета основывается на продуктах с изначально низким уровнем фенилаланина или специально обработанных для лечебного питания.
В некоторых случаях возможно эффективное применение тетрагидробиоптерина, коэнзима фенилаланингидроксилазы, который стимулирует активность фермента и помогает снижать количество фенилаланина.
Помоги своему ребенку
Мозаичный вариант СД, несмотря на более легкое течение, требует постоянной работы с такими детьми. К сожалению, окончательно избавиться от патологии невозможно, но с помощью многопрофильной терапии можно помочь ребенку лучше адаптироваться к социуму и не чувствовать себя обделенным.
Очень важно для детей с синдромом Дауна проводить массаж и лечебную физкультуру. Они позволят повысить тонус мышц и укрепить их, устранят контрактуры, стабилизируют работу суставов и всего опорно-двигательного аппарата
Массаж показан с 2 недель рождения. Но не все приемы позволительно использовать в этом возрасте. Разминание не применяется детям до 3 месяцев. Если у малыша есть проблемы с сердцем, следует проконсультироваться с врачом по поводу массажных приемов.
Из методов ЛФК обязательно используйте упражнения на мяче. Благодаря им происходит развитие двигательных рефлексов и координации.
Показаны для детей с СД занятия плаванием. Оно выполняет функцию гидромассажа.
Особо выделяют такие методы лечения, как иппотерапия и дельфинотерапия.
Иппотерапия – это терапевтическое воздействие, достигаемое при общении с лошадьми. Такой метод обеспечивает:
- развитие равновесия и внимания;
- придает уверенности в себе;
- тепло, которое исходит от лошади, успокаивает ребенка и дает ему чувство защищенности.
Это отличный метод, позволяющий уравновесить нервную систему людей с любым типом синдрома Дауна. Но он требует выполнения некоторых условий:
Важно, чтобы лошадь была большая, а не пони. Они чрезмерно суетливы.
Перед тем как сесть на лошадь, с ней нужно установить контакт: поговорить, покормить, погладить.
Животное должно быть без седла и подков.
Не рекомендуется кататься по асфальту
Дельфинотерапия обеспечивает расслабление и улучшение настроения, придает уверенности в себе, помогает приобрести навыки общения. Этот метод также стимулирует физическое развитие. Ультразвук, который выделяют дельфины, восстанавливает нормальные биотоки.
К другим способам терапии относят:
- назначение медикаментозных средств — гормоны, психостимуляторы, нейрометаболиты, витаминные комплексы;
- консультации психолога и психотерапевта;
- диетотерапия — нередко такие больные страдают ожирением, которое способно приводить к другим патологиям. Поэтому следует соблюдать систему правильного питания, чтобы предотвратить чрезмерный набор веса;
- постоянное наблюдение у узких специалистов.
Диагностика Фенилкетонурии (ФКУ) у детей:
Для диагностики фенилкетонурии (ФКУ) у детей определяют содержание крови уровней фенилаланина и тирозина в крови. Применяют тест Гатри, пробу Феллинга, флуориметрию, хроматографию, МРТ, поиск мутантного гена, электроэнцефалографию.
ЭЭГ позволяет обнаружить нарушения в основном в виде паттерна гипсартимии, даже если приступов у ребенка не наблюдалось. Также находят фокусы спайк- и полиспайк-разрядов (единичные и множественные). МРТ не находит изменений сигнала в стволе, мозжечке или коре головного мозга. Изменения на МРТ не коррелируют с уровнем интеллекта, они зависят от содержания фенилаланина в крови.
Если у ребенка фенилкетонурия II, то симптомы проявляются после 12 месяцев жизни. В крови затем находят повышенный уровень фенилаланина в периоде новорожденности, назначают диету, но болезнь всё равно прогрессирует. У малышей выраженная умственная отсталость, судороги, признаки повышенной возбудимости, гиперрефлексия, мышечная дистония, спастический тетрапарез. Летальный исход в части случаев наступает в возрасте от 2 до 3 лет.
Симптомы фенилкетонурии III напоминают выше перечисленные. У ребенка врачи обнаруживают три типичных признака:
- спастический тетрапарез
- микроцефалия
- глубокая умственная отсталость
Меню для грудного ребенка с ФКУ
в возрасте 2 месяцев с массой тела 5 000г, с индивидуальной толерантностью фенилаланина 175мг/сутки
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 35 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 175 | 12 | 24 | 68 | 540 | |
| Грудное молокоXP Analog LCP 60г + 360мл воды | 390 | 175 | 4.29 | 15.6 | 27.3 | 276.9 |
| 420 | 6.06 | 16.56 | 33.78 | 308.4 | ||
| Суточная норма | 175 | 12,09 | 29,4 | 59,7 | 561,9 |
* Ребёнок может получить:
- 1 способ:
- 3 x 130мл грудного молока и 3 x 120мл XP Analog LCP (1 порция = 20г препарата XP Analog LCP = 4 мерки + 120мл воды)
- 2 способ:
- 6 x 60мл XP Analog LCP (1 порция = 10gг препарата XP Analog LCP = 2 мерки + 60мл воды) и 6 x 60мл грудного молока
| Кол-во (мл) | Фенилаланин (мг) | Белок (г) | Жиры (г) | Углеводы (г) | Энергия (ккал) | |
|---|---|---|---|---|---|---|
| Диетическая норма на кг/сутки | 35 | 2,4 | 4,9 | 13,6 | 108 | |
| Диетическая норма в сутки | 175 | 12 | 24 | 68 | 540 | |
| Грудное молокоMilupa PKU 1-mix (60г + 418мл воды) = | 390 | 175 | 4.29 | 15.6 | 27.3 | 276.9 |
| 420 | 6.06 | 16.56 | 33.78 | 308.4 | ||
| Суточная норма | 175 | 10,35 | 32,16 | 61,08 | 584,4 |
* Ребёнок может получить:
- 1 способ:
- 3 x 130мл грудного молока и 4 x 104мл Milupa PKU 1-mix (1 порция = ???г препарата Milupa PKU 1-mix = 3,5 мерки + 104 мл воды)
- 2способ:
- 7 x 60мл Milupa PKU 1-mix (1 порция = 8,6г препарата Milupa PKU 1-mix = 2 мерки + 60мл воды) и 7 x 56мл грудного молока
















Свежие комментарии